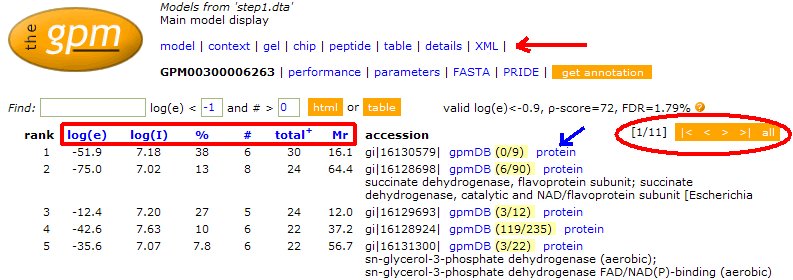
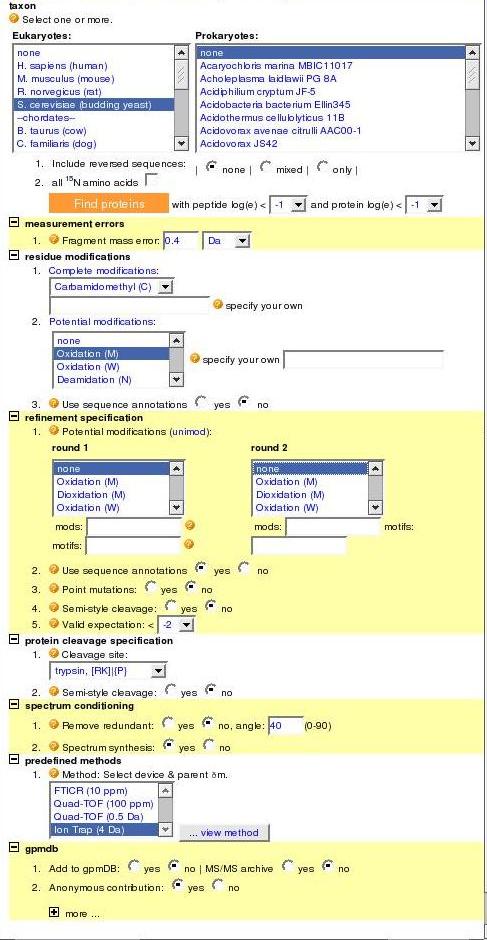
On the right is the main search tool, available from The GPM website when you click on "Genomes". Below, are descriptions of each section
- 1) Click the "Browse" button to upload a file from your computer. We will be using .dta data files linked to this webpage.
- 2) Here you can select one or more organism protein databases to search. For this tutorial, we will use yeast (S. cerevisiae).
Also note that the "Find models" button in this section is used to start your search - But we have to finish putting in our parameters first!
- 3) The measurement error represents how much error we allow in our y- and b- ion calculations. Leave it set to 0.4 Da
- 4) Residue modifications are caused by the chemicals used in preparing our sample. We used iodoacetamide (IAA) to prepare the samples, so
we expect to find carbamidomethylation. We can also specify oxidation of other amino acids, such as methionine, but for the purposes of this
quiz section, you can set all potential modifications to none.
- 5) Set all these modifications to none as well. Set point mutations and semi-style cleavages to "no".
- 6) We used trypsin to digest our sample, so make sure the trypsin cleavage site is selected. Set semi-style cleavage to "no".
- 7) Ignore this section.
- 8) This section specifies what type of mass spectrometer was used. For all the data in this quiz section, we used an "Ion Trap".
- 9) This section allows us to add our results to the GPM database. Since the results are in there already, you can set the
anonymous contribution to "yes" and all other fields to "no".
|
|