craw_conf.py Documentation
Overview
Crawdad consists of a sequentially applied set of modules, as shown in the flowchart below 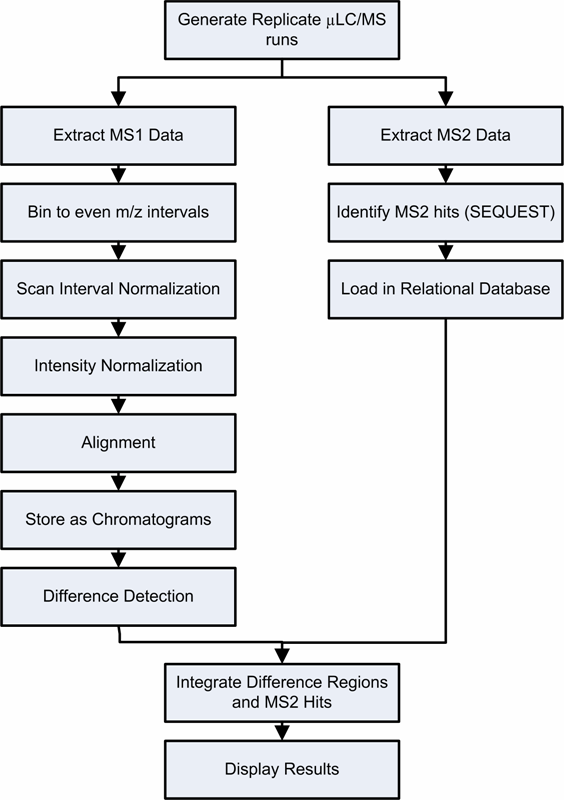
Briefly, the steps consist of:
- Extracting ms1, ms2 data -- this is done with MakeMS2 on the instrument machines. .ms1 and .ms2 files are then moved to proteome
- Binning to m/z intervals -- done with ms1_to_msmat.py script (TODO -- merge into MakeMS2)
- Scan interval normalization -- scans are resampled so as to be at a consistent timescale
- Chromatogram Smoothing -- chromatograms are smoothed using a savitsky-golay smoothing window
- Alignment -- each run is aligned in the retention time dimension to a common master template run
- Detecting Differences -- use the t-test on aligned data to find regions of differential intensity
- Extraction of MS/MS data, SEQUEST, etc... (use hermie)
- Associate MS/MS peptide IDs with difference regions -- you load your sqts into a database, as well as your results
from difference detection, and produce a set of annotated differences
- Assemble difference regions that are annotated with peptide identifications into lists of proteins and peptides
that are changing in abundance
All these steps are controlled through the craw_conf.py application
.
craw_conf.py is a control script for CRAWDAD which uses an XML configuration
file to define the steps and parameters for pre-processing, aligning, discovering signal differences, and mapping those differences to peptide identifications. Most interactions with craw_conf.py are set in the align_config.xml file, which is outlined below
align_config.xml
A sample align_config.xml file is shown here
the steps, or 'actions' as listed above are given a label in align_config.xml, and output
for those steps are saved to the directories named after the labels. If the computer crawdad is running is able to produce images w/ python, then base peak chromatograms will be produced as images from the output of each sample group for each step.
these are.
Generally speaking, if you edit the config file to run jobs on the queue,
(using the ) as shown above, craw_conf.py will
submit one single-processor job per file per step as outlined above. You can process more
than two 'sample groups' at once, but at the moment comparisons to find differences are limited
to comparing two.
run visualization programs
We have two classes of run visualization programs:
2D-heatmap based views of an entire LC-MS run
msmat_img.py will display an LC-MS run from an .msmat file as a 2D heatmap, which is zoomable.
Instructions are available
chromatogram / spectra viewing software
chrom_viewer.py displays one or more spectra or chromatograms (base peak, XIC, or TIC)
Instructions are available
these programs are useful for viewing individual steps of the output, i.e. for
assessing alignment quality, or determining if the appropriate smoothing has been used
CRAWDAD File
MSMAT format files Store the initial binned MS1 data and intermediate steps of individual runs
End Products from CRAWDAD
brief notes on XML